
RESEARCH ARTICLE
In Vivo Validation of a Bioinformatics Based Tool to Identify Reduced Replication Capacity in HIV-1
Christina M.R Kitchen*, 1, 2, Paul Krogstad 2, 3, Scott G Kitchen 2, 4
Article Information
Identifiers and Pagination:
Year: 2010Volume: 4
First Page: 225
Last Page: 232
Publisher Id: TOMINFOJ-4-225
DOI: 10.2174/1874431101004010225
Article History:
Received Date: 12/3/2010Revision Received Date: 11/6/2010
Acceptance Date: 29/8/2010
Electronic publication date: 3/12/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Although antiretroviral drug resistance is common in treated HIV infected individuals, it is not a consistent indicator of HIV morbidity and mortality. To the contrary, HIV resistance-associated mutations may lead to changes in viral fitness that are beneficial to infected individuals. Using a bioinformatics-based model to assess the effects of numerous drug resistance mutations, we determined that the D30N mutation in HIV-1 protease had the largest decrease in replication capacity among known protease resistance mutations. To test this in silico result in an in vivo environment, we constructed several drug-resistant mutant HIV-1 strains and compared their relative fitness utilizing the SCID-hu mouse model. We found HIV-1 containing the D30N mutation had a significant defect in vivo, showing impaired replication kinetics and a decreased ability to deplete CD4+ thymocytes, compared to the wild-type or virus without the D30N mutation. In comparison, virus containing the M184V mutation in reverse transcriptase, which shows decreased replication capacity in vitro, did not have an effect on viral fitness in vivo. Thus, in this study we have verified an in silico bioinformatics result with a biological assessment to identify a unique mutation in HIV-1 that has a significant fitness defect in vivo.
INTRODUCTION
The rate of HIV-1 replication and mutation in infected individuals is remarkable and alarming [1-3]. During antiretroviral treatment there is a dynamic interplay of drug-resistance and fitness occurring within the virus population. It has been proposed that reduced viral fitness of HIV-1 can result in slower disease progression in vivo due to the decreased pathogenic ability of the virus [4-6]. Viral fitness in this case is defined by the ability of a virus to replicate in vivo and produce pathologic changes in host tissues. While it is difficult to directly assess viral fitness in vivo, the relative replication capacity (RC) of a viral mutant may be determined using in vitro cell culture models by examining how the virus replicates when compared to a virus without any mutations (wild-type virus) [4, 7, 8]. Viral fitness in vivo and observed RC in vitro are clearly linked; however, there is a lack of direct evidence that certain drug-resistance mutations actually confer a defect in viral fitness in vivo in infected persons.
In the clinical setting, combination antiretroviral therapy (ART) often fails to completely and durably suppress plasma levels of HIV-1 RNA [9-12] and the viral load may rebound. Virologic failure is often a consequence of a series of mutations in HIV that decreases the susceptibility of the virus to antiretroviral agents. It has been shown that drug-resistant HIV often has lower RC in vitro than wild-type virus [4, 7, 8, 13]. Treatment interruption of ART in patients leads to the re-emergence of archived wild-type virus that has a higher RC than the circulating drug-resistant virus and is associated with higher viremia and decreased CD4 T-cell counts [14, 15]. In contrast, patients who remain on their ART regimen despite ongoing viral replication may have stable CD4 counts and stable viremia [5, 16]. In one report, only 36.8% of patients experienced a decrease in CD4 counts to pre-therapy levels while remaining on a failing ART regimen for 3 years [16]. Similarly, Barbour et al. [17] found patients who remained on a failing regimen had stable RC, viremia and CD4 counts. These studies suggest that HIV is subject to genetic bottlenecks where it cannot create further resistance without sacrificing its ability to replicate.
Decreases in viral RC due to mutation have been well-characterized in vitro, although alterations in fitness in vivo is often correlative due to the lack of use of a controlled experimental system during these studies [4, 7, 8, 13, 17-20]. It has been shown that certain mutations cause a substantial decrease in RC in vitro relative to wild-type strains. However, there also exist mutants that have similar or even higher replication capacity than wild-type [21]. The D30N mutation in the protease (PR) gene involves a GAT to AAT mutation that is well-characterized and specific to the protease inhibitor nelfinavir (NFV) [22, 23]. The D30N has been identified to biochemically alter viral protease activity in heterologous cleavage studies [7, 13, 18, 23-25]. The M184V mutation in the reverse transcriptase (RT) gene, which causes primary resistance to lamuvidine (3TC), has been extensively studied and is common in treated patients [6, 26-28]. Viral isolates with the M184V mutation in RT have lower RC than viral isolates without the mutation [17]. In one study, HIV infected individuals with significant ART drug resistance were randomized to receive 3TC or to stop ART completely; those receiving 3TC alone experienced a lower increase in viral load, a slower rate of decline in CD4 T cell percentage, and fewer adverse clinical events related to HIV infection [26].
Assessing the relative impact of mutations on HIV fitness is difficult because there are often many more parameters than there are data. Previously we described an informatics-based method to identify the relative fitness cost of mutations in protease using a Bayesian hierarchical model [29]. Using this model we found mutations in protease that had a relatively large decrease in viral RC. However, it is unknown if the in silico results correspond to an actual in vivo fitness decrease.
The effect of mutations on viral fitness and pathogenicity often cannot be determined in vitro; however, the SCID-hu mouse model allows direct assessment in human tissue in an in vivo setting. The SCID-hu mouse model of HIV infection is well described and has been used to examine the mechanisms of viral pathogenesis in primary human lymphoid tissue [8, 30]. We and others have found that HIV-1 directly injected into human thy/liv implants in SCID-hu mice results in reproducible infection and severe depletion of human cells bearing the CD4 molecule [30-32]. Stoddart et al. [8] examined viral fitness in 8 clinical isolates compared with HIVNL4-3, however none of these isolates contained the D30N mutation. They determined that while the RC of PI-resistant strains of HIV-1 in peripheral blood mononuclear cells (PBMCs) was moderately impaired compared to wild-type (WT) virus, the RC of PI-resistant strains in vivo was highly impaired. The observation that differences in replication between PI-resistant strains and WT stains were seen in vivo in the SCID-hu mouse and not in vitro in PBMCs indicates that the SCID-hu mouse is an excellent model system to assess the fitness of viruses with different antiretroviral drug resistance mutations in primary human tissue. In contrast to other studies with patient isolates [8, 33], we explicitly tested specific mutations found a priori to have a fitness effect. In the current study, we utilized results from a bio-informatics model and then utilized in vitro tissue culture and the in vivo SCID-hu mouse model to determine if this mutation confers a biological fitness defect. This study demonstrates the synergistic possibilities in translational research and validates a unique drug resistance mutation in HIV-1 that confers an in vivo fitness defect in the virus.
METHODS
Bioinformatics Analysis
We used a Bayesian hierarchical model to determine the relative replication capacity effect of mutations in 161 genotype-phenotype pairs of HIV-1 protease (described in [29]). Because we have a much greater number of parameters than data points (large p, small n), prior specification is critically important. Fortunately, there exists a wealth of information elucidating links between HIV replication and specific mutations based on site-directed mutagenesis and drug-resistance studies. Bayesian methods allow us to explicitly take into account what is known a priori about mutational patterns in HIV-1. The idea is to provide priors with both shrinkage and variable selection components. Let yi be the continuous fitness phenotype for the ith HIV-1 sequence ( Y = (y1,..., yn ) ' ). Let xij represent the jth codon position for the ith sequence ( Xj = (x1j , ..., xnj ) ' ). In this case the xj’s are 0/1 indicators of a mutation away from wild-type at each codon position along the protease genome. The informatics problem is to find the set of Xj’s that are contributing to the fitness phenotype. Because the parameter space is much larger than the number of sequences, backwards and forward selection using conventional regression methods are unlikely to yield useful results. Kuo and Mallik [34] created a class of prior that has both a shrinkage and a selection component where all coefficients are exchangeable. Following Kuo and Mallik the prior parameters are specified by assuming all regression coefficients have the same prior. There are two parts to this prior, the δj is the model selection part and the prior on the regression coefficient βj is the shrinkage component. βj , the prior mean and variance of the regression coefficient, is modeled as a Normal with hyperpriors on the mean and variance which are fixed. The δj , (δj ~ Bernoulli(ρ)) , is a binary indicator function for the presence of a fitness effect on the regression coefficient, (which then becomes βjδj ) in the model. This prior is the same for all regression coefficients and thus all of the xij are exchangeable. Fig. (1A) illustrates this prior. This model is scientifically uninformative in that each codon position has the same prior effect on fitness. We call this Model 1, the uninformative prior.
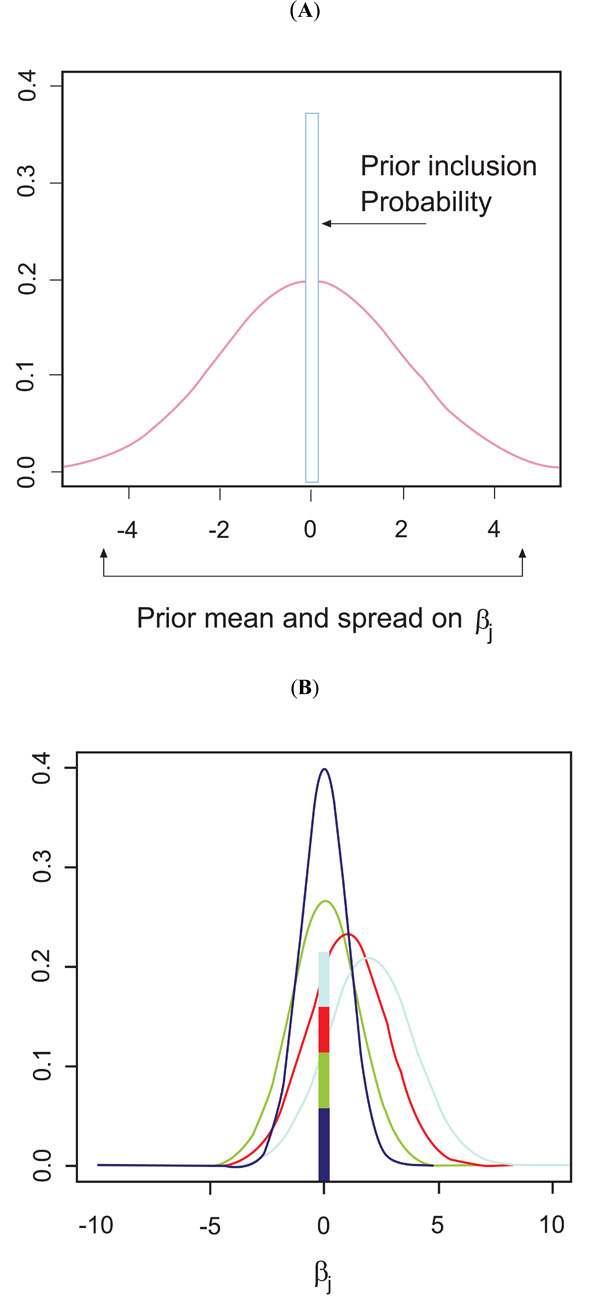 |
Fig. (1). Prior illustration for the models. For each codon position j, the column shows δj, the prior 0/1 indicator if codon j is important to the fitness phenotype. The curve shows the prior distribution on the regression coefficient βj. (A) Model 1, the uninformative prior. All codon positions have the same prior. (B) The Exchangeable on Subsets Prior (ESP). In the figure, 4 subsets are shown. Each codon within a subset has the same prior δj and prior distribution on βj. Different subsets have different values for the priors on the δj’s and βj’s. The four subsets are shown in different colors. |
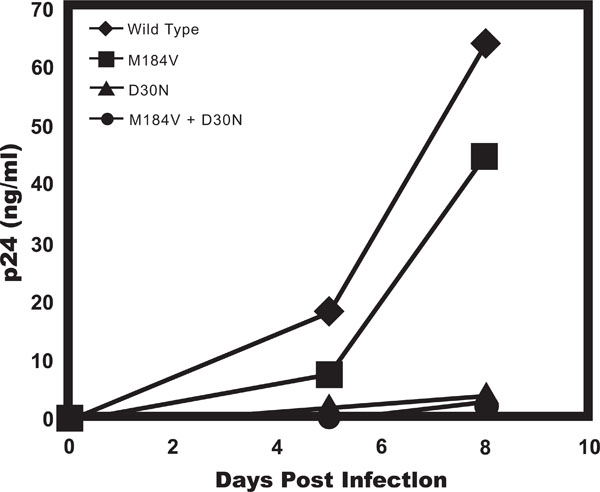 |
Fig. (2). Replication kinetics of wild-type, M184V-containing, D30N-containing, and M184V and D30N-containing viruses in PHA-stimulated PBMC. Viral p24 antigen production was assessed at days 5 and 8 post infection. The results shown are representative of 3 independent experiments with PBMC derived from different donors. |
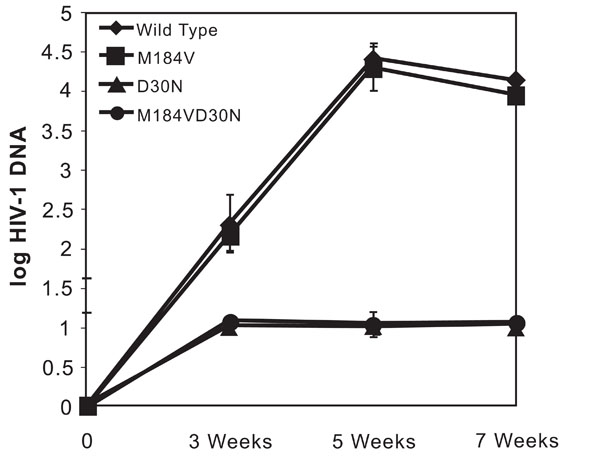 |
Fig. (3). Viral replication of wild-type, M184V-containing, D30N-containing, and M184V and D30N-containing viruses in vivo. Thy/liv implants were infected, separately (n=5 mice per group), with identical amounts of infectious units of each virus. Implants were analyzed following biopsy of infected tissue by quantitative PCR for HIV proviral DNA at 3, 5, and 7 weeks following infection. The amounts of proviral DNA are provided as copies of full length proviral DNA per 100,000 cells, as determined by the use of HIV and human β-globin specific primers and quantitative comparison to known controls. Mock infected controls were negative for HIV proviral DNA (not shown). |
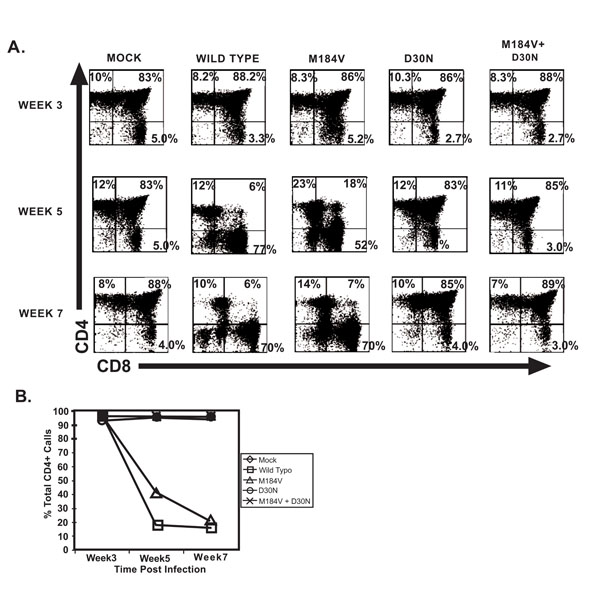 |
Fig. (4). Depletion of CD4-bearing cells following infection with wild-type, M184V-containing, D30N-containing, and M184V and D30N-containing viruses in vivo. (A) CD4 versus CD8 flow cytometry staining profiles were determined on biopsied thy/liv tissue at the indicated times following infection with one of the four indicated viruses. Cells were stained with monoclonal antibodies (and their corresponding fluorochromes) to CD45 (FITC), CD8 (ECD), and CD4 (APC). CD4 and CD8 profiles of this population are given by gating on the human CD45+ population of cells. The percentages of cells in each quadrant are indicated. The data is representative of one mouse per group (total of 5 mice per group) of a total of three experiments. (B) Graphic representation of the percentage of total CD4+ cells in the respective thy/liv implants over time. |
Site-directed mutagenesis and HIV-1 drug-resistance studies have gathered a wealth of information on identifying codon positions that appear to have an in vitro effect on fitness. To incorporate this information into our model we generalize our KM priors to allow for subsets of codon positions whose effects are exchangeable within the subset (the Exchangeable on Subsets Prior (ESP)). That is codons within the same set have the same prior while codons in different subsets have different priors. To see how this might work, we could allow a set of codons that site-directed mutagenesis has implicated as having a fitness effect to have a higher probability of inclusion (higher ρ ) and a higher prior mean fitness effect (through the mean and variance hyperparameters of βj) that codons not in this set. All of the codons in this selected subset are exchangeable, i.e. have the same prior. However, codons that are not in this subset could have a prior δj that has a lower probability of inclusion, ρ ,and a smaller fitness effect, βj . Fig. (1B) illustrates this ESP prior for when there are 4 different subsets. However the scientific literature uses different methods to determine importance and although there is general agreement for certain codon positions, there is not a consensus for all positions. Because of this, we create 3 ESP priors based on 3 relevant papers in the HIV-1 literature.
Specifically, we created a prior based on the paper by Swanstrom and Erona [35], a prior based on the paper by Foulkes and DeGruttola [36], and one based on Loeb et al. [18]. For each paper a model is constructed in which all codon positions identified as having a fitness effect are deemed important. These codon positions are all given the same prior with high inclusion probability and higher prior fitness effects. Codons that are not identified in the paper are deemed unimportant and are all given priors with low values of rho and diffuse βj’s centered around zero. To assess the consensus of positions we created another ESP prior where each model (the 3 literature based models and the uninformative Model 1) “voted” for a specific codon to be included in the “important” set and that codon was weighted according to the number of votes. In the voting model there were 4 classes of priors according to the number of votes a codon position received: 0, 1, 2, 3 or 4. Codons with more votes had a higher probability of inclusion, and beta coefficients with larger fitness effects.
To choose which prior fit the data best we used Prior Model Selection (PMS). In brief, we used Gibbs sampling to generate draws from the posterior distribution f (β ,δ | y) of each of the 5 models. The marginal likelihood of Y, m(y) = f (y | θ )π (θ) / π (θ | y) , was calculated using Chib’s [37] method; where θ represents the parameters and π represents the prior. Each model was run 10 times to obtain an estimate of the standard deviation of the log marginal likelihood for each model. Bayes Factors were calculated from the difference of the average log marginal likelihood values and were used to compare the models. To determine the in vivo effect in the SCID-hu mouse model, we conducted a power analysis to determine the number of mice that would be required in each group. The study was powered to detect a difference in the D30N groups (D30N and D30N + M184V) versus control (wild-type). The effect size of the power calculations were based on the results of the statistical model presented in Kitchen et al. [29]. For each mouse, the cumulative area under the curve for log HIV-1 RNA viral burden and total CD4+ thymocyte count were calculated. Groups were compared using the two-sided Wilcoxon Rank Sum Test. Groups were also compared over time using a nonlinear mixed effects model. Group by time comparisons were made using the Wilcoxon Rank Sum test with p-values adjusted for the overall type 1 error rate using a Bonferroni correction.
Generation of Drug-Resistant Viral Stock
Viral mutants were generated by the introduction of the D30N and M184V mutations into a molecularly cloned strain of HIV (HIV-1NL4-3) by site-directed mutagenesis. Viral stocks were prepared by electroporation of CEMx174 cells with plasmid DNA encoding the genome of wild-type and mutant viruses. Virus was harvested in culture supernatant 2 and 3 days following electroporation and quantitation of p24 gag was performed by enzyme-linked immunosorbent assay (ELISA, Coulter, Hialeah, Florida). Viral titers were determined through a standard limiting dilution assay on CEMx174 cells.
In Vitro Viral Growth
Freshly isolated peripheral blood mononuclear cells (PBMC) were obtained in the form of leukopacks from anonymous donors by the UCLA AIDS Institute Virology Core in accordance with IRB protocols. PBMCs were purified by Ficol purification and cells were then stimulated for three days with phytohemagglutin (PHA)(1 microgram/ml)(Sigma) and IL-2 (100 units/ml)(R&D Systems, Minneapolis, MN). Cells were then infected, separately, with each indicated virus at a multiplicity of infection (MOI) of 0.003 in a volume of 1 ml for two hours at 37 degrees. Cells were then cultured at a concentration of 1 x 106/ml and at the indicated times 100 microliters of supernatant was removed and replaced with fresh medium. Viral supernatant was then placed in PBS containing 1% Triton X-100 and p24 levels were quantitated by ELISA, as described above.
SCID-hu Mice
SCID-hu thy/liv mice were constructed by implanting human fetal thymus and liver under the kidney capsule of C.B.17 SCID mice as described [28, 34]. Thy/liv implants (n=5 mice per group) were infected by direct injection of 100 infectious units of either wild type HIV-1NL4-3 or HIV-1 NL4-3 containing the D30N, the M184V, or both the D30N and M184V mutations or were mock infected with medium alone. At the specified time points, thy/live implants were biopsied utilizing survival surgery procedures as described [28, 34].
Flow Cytometry
Single cell suspensions were made from the biopsied tissue and analyzed by flow cytometry for the expression of CD45, CD3, CD4, and CD8 similar to that described [35, 36]. Cells were run on a Coulter FC500 (Coulter, Hialeah, FL) flow cytometry and data was analyzed by FlowJo software (Treestar, Ashland, OR). Depletion of CD4+ cells (CD4+CD8+ and CD4+CD8- thymocytes) was determined by comparison of cells from HIV infected implants to mock-infected controls.
Quantitative DNA PCR
A fraction of cells was removed and DNA was purified as previously described [38, 39]. Quantitation of proviral and cellular DNA was performed with real time quantitative PCR using primers specific for human beta globin sequences and full-length HIV reverse transcripts (the long terminal repeat-gag junction) as described by [40].
RESULTS
Utilizing a data set of 161 of genotype/phenotype pairs (described in [29]), we used a Bayesian hierarchical model to determine the relative effects of mutation in silico. The priors that were used included an uninformative prior across the whole protease genome, three literature based ESP priors and a voting ESP prior whereupon each model “voted” for a codon position. We then incorporated prior model selection (PMS) to choose among our priors. The voting prior had the smallest log marginal likelihood of all the models and testing the uninformative model versus the voting model decisively rejected the uninformative model (log Bayes Factor=91.32). Using this model as our final model, we were able to assess the relative cost of each mutation in protease, in terms of RC. The model found that the D30N mutation had the largest decrease in RC relative to other resistance-associated mutations suggesting that this mutation may have clinical benefit in prolonging disease progression by conferring a RC defect. Table 1 lists the regression coefficient estimates and the 95% credible interval of the top 5 drug-resistance associated mutations in protease.
The Estimated Fitness Effect, E[βδ|γ] and the Corresponding 95 Percent Credible Interval for the Top 5 Drug-Resistance Associated Mutations from the Final Best Fitting Model
| Codon Position | Estimated Fitness Effect | 95% Credible Interval |
|---|---|---|
| D30N | -16.72 | (-30.32, -2.92) |
| V77I | -13.49 | (-22.34, -4.62) |
| V82A/F/T/S | -16.61 | (-25.40, -5.35) |
| I84V/A/C | -13.11 | (-23.28, -0.81) |
| L90M | -12.60 | (-23.25, -1.91) |
To assess the effects the in silico identified D30N PR and the previously implicated M184V RT mutation in infectivity of HIV-1 we assessed viral titers of molecularly clones variants of HIV-1NL4-3 containing these mutations in a standard limiting dilution assay. HIV-1NL4-3 wild type, HIV-1NL4-3 with the D30N mutation, HIV-1NL4-3 with the M184V mutation, and HIV-1NL4-3 containing both the D30N and M184V mutations all had a titer of 300 picograms per infectious unit, indicating that the presence of the mutation in the drug resistant viruses did not affect initial viral infectivity. We then compared the ability of theses viruses to replicate in PHA-activated PBMCs. Cells were infected at a relatively low multiplicity of infection (MOI) to detect and magnify differences in the ability of the virus to replicate over multiple rounds of infection. We found that the virus containing the M184V mutation displayed slightly delayed replication kinetics while the viruses containing the D30N alone and in combination with M184V and the double mutant had a more dramatic decrease in viral replication as compared to the wild-type virus (Fig. 2). In all, these data indicate that mutations do not alter the infectivity of the virus, but reduce viral RC.
To examine the effects of these mutations on viral fitness and pathogenesis in vivo, we examined the accumulation of HIV DNA and the level of virus-induced CD4+ thymocyte depletion over time using the SCID-hu mouse model. Thy/liv implants were initially infected with equivalent amounts of infectious units of wild type, D30N-containing, M184V-containing, and both D30N and M184V containing HIV-1NL4-3 in parallel and the effects of the virus were examined at 3, 5, and 7 weeks following infection. Proviral DNA was detected in mice infected with each different virus within three weeks post infection (Fig. 3). Infection of thy/liv implants with wild type (NL4-3) produced time dependent increases in log HIV-1 DNA levels. Infection of mice with HIV-1NL4-3 containing the M184V mutation produced similar viral kinetics as the wild type virus. The average cumulative area under the curve was not significantly different for M184V versus wildtype. Mice infected with HIV-1NL4-3 containing the D30N or D30N+M184V mutation, however, had barely detectable viral DNA at all three time points. Mice harboring HIV-1NL4-3 containing the D30N mutant strains had significantly lower levels of HIV RNA than the wildtype or M184V alone groups (P=0.001). There was no statistically significant difference in HIV DNA levels between the D30N and the D30N+M184V infected group at any time point.
Cellular depletion of CD4 bearing thymocytes which is indicative of viral pathogenesis, primarily in the CD4+CD8+ population, was observed in mice infected with wild type HIV-1 NL4-3 and the M184V containing strains within 5 weeks following infection (Fig. 4). Seven weeks post inoculation, we found profound CD4+ thymocyte depletion in the mice infected with wild-type virus or the virus containing the M184V mutation alone. The level of depletion in these two groups was significantly greater than the level of depletion found in mice infected with strains that included the D30N mutation (p=0.001). In fact, mice infected with strains containing the D30N mutation (including the double mutant) were not statistically different from the mock infected mice in terms of percent total CD4+ thymocytes. The differences in percent total thymocytes between mock and the two D30N groups were not significant at any time point (even without adjusting for multiple comparisons). Mice infected with the M184V mutation alone had a profound depletion in thymocytes compared to mock infected mice (p=0.027) and was not significantly different from the depletion found in mice infected with wildtype virus, indicating that the D30N is primarily responsible for the attenuated virulence.
DISCUSSION
These results illustrate the utility of biostatisticians and biologists working together and demonstrate the synergy possible with translational studies. This work was prefaced by a thorough examination of protease mutations in a bioinformatics system whereby the D30N mutation was found to have a profound effect on in vitro replication. To be able to specifically attribute viral attenuation with the mutation, we constructed point mutants instead of using patient isolates that may have accrued mutations at other loci in the viral genome. Our results indicate that the D30N mutation has a substantial effect on the ability of HIV-1 to deplete thymocytes and to replicate in an in vivo system.
Our data clearly demonstrate that mutations in the HIV-1 genome may differ greatly in their impact on HIV replication capacity and pathogenicity. Whereas the virus containing the M184V mutation alone conferred a RC defect in vitro, we did not find evidence for decreased pathogenicity of this virus in vivo. In our experiments, mice infected with this mutant did not have statistically different viral loads or CD4+ thymocyte counts than mice infected with wild-type. In addition, the M184V did not enhance or inhibit viral replication or the ability of the virus to deplete thymocytes when coupled with the D30N mutation. There was no statistically significant difference between mice infected with the D30N mutant and those infected with the D30N+M184V double mutant at any time point. These studies demonstrate that RC results observed in vitro do not necessarily correlate with a defect in viral fitness in vivo.
Our results contrast from those described by Stoddard et al. who found that mice infected with a virus (210P) containing protease mutation at I54V and V82A had a higher level of viremia than mice infected with wild-type virus. However, comparison is difficult due to the presence of different mutations in the HIV protease (D30N versus I54V+V82A), as well as the variability inherent in experiments employing SCID-hu mice in which a chimeric organ is created by engraftment of primary human tissues. Nonetheless, the differences in these results may also suggest that virus containing the D30N mutation is less able to replicate in the thymus than virus containing the V82A mutation (as is suggested by our bioinformatics model). The D30N mutation was chosen as it had the largest decrease in relative replication capacity. It is noteworthy that previous studies have shown that the V28A and the D30N mutations do no co-occur in the same genome [41-43].
Although some patients experience the so called “discordant state” characterized by having both high viral loads and stable CD4+ T-cell counts, but there are many others who are concordant and have high viral loads and decreasing CD4 counts despite the presence of PI resistance mutations [5, 16, 44-46]. The latter state likely arises when HIV acquires compensatory mutations that increase its level of fitness and restore its ability to deplete CD4+ T-cells. The apparent lack of pathogenicity of the HIV-1 D30N mutant suggests that this mutation may represent a genetic “dead-end” for the virus. Once the D30N mutation has been acquired, the virus may not be able to replicate well enough to return to wild-type fitness. There may be other genetic bottlenecks in the virus that can be capitalized upon to drive the virus into an unfit-state and preserve CD4+ T-cells, however further research is needed.
In people infected with clade B strains of HIV, those that harbor virus containing the D30N mutation often also have the L63P mutation. The L63P mutation was not identified as having a large fitness defect by our model, likely because the L63P mutation is prevalent in patients who are treatment naïve and is therefore not a drug-resistance mutation. Moreover, others have reported that the replication of a D30N containing variant was not significantly different from a L63P+D30N dual mutant [7]. Similarly, patients with the D30N mutation often develop the N88D/S mutation. N88D/S did not have a significant effect on fitness in the statistical model and was not tested. HIV variants with both the D30N and N88D mutation was also found to have decreased replication capacity in vitro, including subtype C strains [24, 47, 48].
There are several limitations to this study. Our analysis was based on examinations of mutations found in protease in HIV-1 clade B strains and our biologic experiments used viruses based on an HIV-1 clade B strain. Although the D30N has been identified as having an effect in vitro in HIV-1 clade C strains, it not known if the in vivo result is generalizable to other non-B clades and HIV-2. It is also possible that the D30N mutation might not be preferentially selected through nelfinavir treatment in non clade-B strains. Further there could be unaccounted variation in the human tissues used in the SCID-hu model that limits the generalizability of this finding. Further research needs to be done.
In conclusion, our results suggest the possibility of the existence of genetic bottlenecks in HIV-1 that select for mutations that diminish replication capacity and in vivo fitness to such an extent that the virus is unable to acquire compensatory mutations as well as deplete target cells. Further work is needed to find other possible dead-end mutations in regions of the genome that are targeted by other antiretroviral agents now under development or in initial use, such as inhibitors of the strand transfer activity of the HIV integrase protein. In all, this study validates a bioinformatics model tested by an in vivo system.
ACKNOWLEDGEMENTS
This work was supported by the Center for AIDS Research UCLA AI07 (CMRK and SK) and the NIAID to PK (AI01996). Effort by PK was also supported by an Elizabeth Glaser Pediatric AIDS Foundation Scientist Award.
