
RESEARCH ARTICLE
Characterization of Chromosomal Translocation Breakpoint Sequences in Solid Tumours: “An In Silico Analysis”
Aditi Daga 1, §, Afzal Ansari 2, §, Rakesh Rawal*, 3, Valentina Umrania 1
Article Information
Identifiers and Pagination:
Year: 2015Volume: 9
First Page: 1
Last Page: 8
Publisher Id: TOMINFOJ-9-1
DOI: 10.2174/1874431101509010001
Article History:
Received Date: 15/12/2014Revision Received Date: 19/2/2015
Acceptance Date: 28/2/2015
Electronic publication date: 30/4/2015
Collection year: 2015
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Chromosomal translocations that results in formation and activation of fusion oncogenes are observed in numerous solid malignancies since years back. Expression of fusion kinases in these cancers drives the initiation & progression that ultimately leads to tumour development and thus comes out to be clinically imperative in terms of diagnosis and treatment of cancer. Nonetheless, molecular mechanisms beneath these translocations remained unexplored consequently limiting our knowledge of carcinogenesis and hence is the current field where further research is required. The issue of prime focus is the precision with which the chromosomes breaks and reunites within genome. Characterization of Genomic sequences located at Breakpoint region may direct us towards the thorough understanding of mechanism leading to chromosomal rearrangement. A unique computational multi-parametric analysis was performed for characterization of genomic sequence within and around breakpoint region. This study turns out to be novel as it reveals the occurrence of Segmental Duplications flanking the breakpoints of all translocation. Breakpoint Islands were also investigated for the presence of other intricate genomic architecture and various physico-chemical parameters. Our study particularly highlights the probable role of SDs and specific genomic features in precise chromosomal breakage. Additionally, it pinpoints the potential features that may be significant for double-strand breaks leading to chromosomal rearrangements.
INTRODUCTION
Chromosomal translocations are recognized as one of the chief cause of tumour progression at molecular level that consequently develops gene fusions (Fig. 1) [1-3]. These fusion genes are ideal prognostic, diagnostic markers and therapeutic targets as they attribute distinct features to specific cancer subtypes. According to earlier presumptions translocations were thought curbed primarily to hematological tumours but recent findings proposes their widespread and rising number characterizing the subset of frequent & rare solid cancers like lung, prostate, kidney, papillary thyroid carcinoma, salivary gland tumours [4-6].
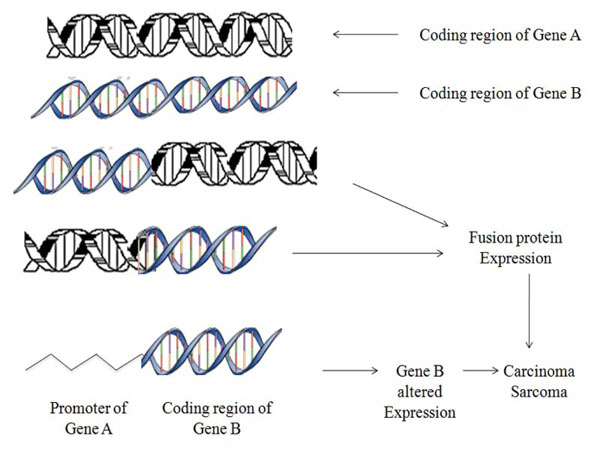 |
Fig. (1). Schematic representation showing mechanism of aberrant chromosomal translocations. |
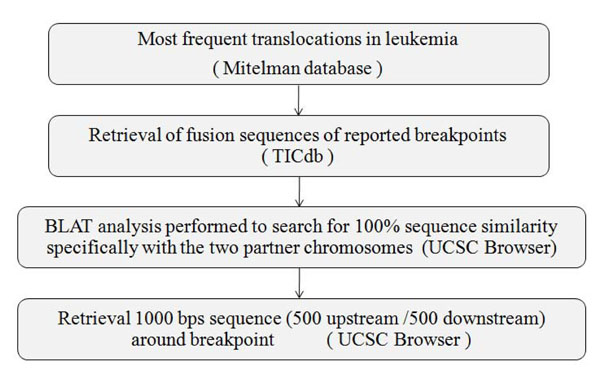 |
Fig. (2). Flow chart for retrieval of 1000 Base pair nucleotide sequences of fusion partners of major translocations from TICdb and UCSC tool. |
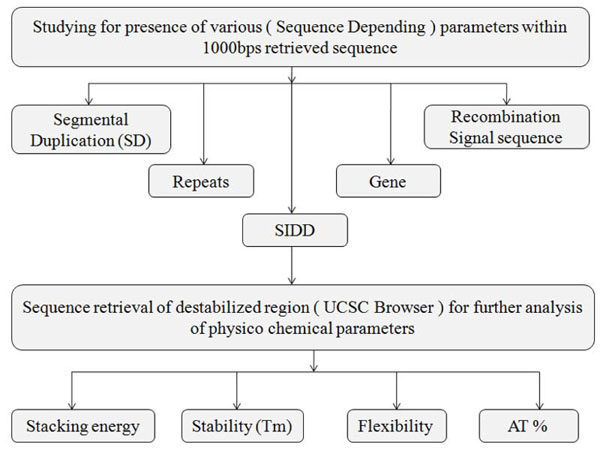 |
Fig. (3). Flowchart showing multiparametric computational analysis for known breakpoint region. |
 |
Fig. (4). Identification of nearest Segmental Duplicons (SDs) by UCSC genome browser for t(21:22). This figure depicts the occurrence of SD at the genomic location (48100446-48117693) of chr 21(A) which matches with genomic segment (51226859-51244566) of chr 22 (B). |
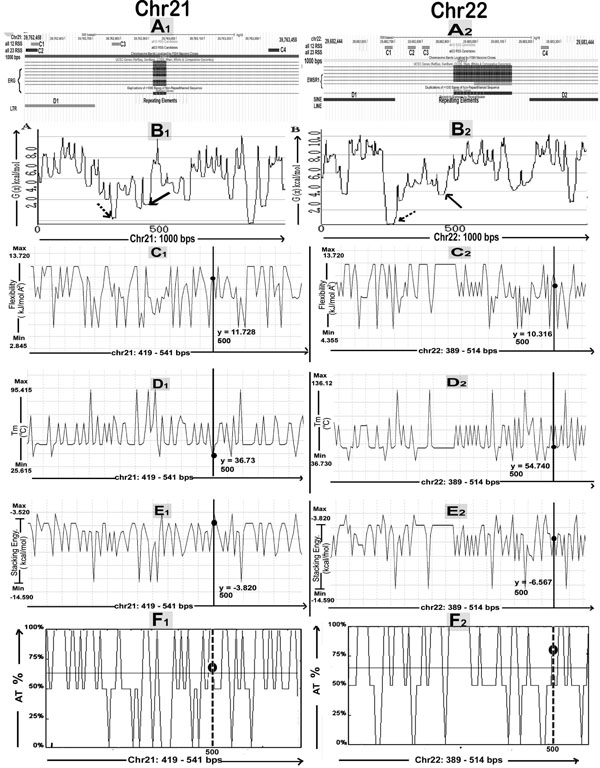 |
Fig. (5). Analysis of sequence motifs, destabilization sites and physico-chemical parametersin BpI region of t(21;22). (A1 & 2) It depicts occurrence of 12 & 23 bps cryptic RSSs and repetitive elements found in the BpI region comprising of the non-coding portion of the respective genes. (B1 & 2) Graphical representation of SIDD destabilization profile plotted for G(x), the incremental free energy against queried sequence. The bold arrow portrays the presence of destabilization site that is in the closest proximity to the breakpoint while the dotted arrow represents SIDD region with the highest destabilization which requires minimum energy for duplex separation. (C1 &2) Flexibility of query sequence was graphically plotted in which the line defines the high helical flexibility at the breakpoint junction (D1 &2) Graphical representation of melting temperature of query sequence was designed. The line demarcates the decreasing trend of Tm at the breakpoint junction. (E1 & 2) Higher energy levels of base stacking was found at the breakpoint junction that is denoted by a line in the pictorial representation (F1 & 2): DNA base composition analysis for SIDD region showed highest AT content at the breakpoint junction, shown by dotted line. |
Like in leukemia, oncogenic fusions in epithelial cancers can be categorized into two broad group (a) tyrosine kinase e.g. papillary thyroid cancers have been characterize by RET fusion. ALK & ROS1 fusions are frequent in NSCLCs and (b) transcription factor e.g. ETV6/NTRK3 fusions are expressed mainly in secretory breast cancer While papillary thyroid carcinomas are distinguishably associated with RET and NTRK1 rearrangements. While in addition, fusions related to TMPSSR2, TFE3, PLAG1, HMGA2 are manifested with occurrence of prostate, renal, salivary gland pleiomorphic adenoma respectively [7-10] (Fig. F1). Assessment of these rearrangements which directs the formation of breakpoints has divulged numerous recurring thoughts providing crucial depth into mechanism of carcinogenesis.
The issue of prime focus is the precision with which the chromosomes breaks & reunites within genome and in order to achieve this, one key feature of particular importance is to study the genomic sequence lying in the vicinity of breakpoints. This will explicate probable role of the genomic architecture and will also define what those potential feature may be. For this, functional annotation are required to be accompanied with physical information to understand the structure, dynamics and the common functionality of genomic DNA, due to which prevalence of breakpoints may be associated with several genomic features.
From previous records it has been emphasized that chromatin structural elements are associated with and responsible for double strand breaks within genome which are incorrectly ligated resulting into recurring chromosomal translocation [11-13]. Therefore, in order to attain profound knowledge of the molecular mechanism of carcinogenesis and a holistic idea concerning the behavioral patterns of these breakpoints, we contemplated to scrutinize a variety of factors including segmental duplicons (SDs), destabilization profiles; Recombination signal sequences (RSS), repeats, physico-chemical characteristics of nucleic acid and many more.
MATERIALS & METHODS
Data Retrieval
Manual curation of chromosomal translocation data was done from Mitelman Database (http://cgap.nci.nih.gov/Chro mosomes/Mitelman) [14] (Fig. 2). The frequently reported translocations leading to formation of oncogenic fusion transcripts were further investigated for involvement of genes in both the partner chromosomes and their fusion sequences were retrieved from TICdb (www.unav.es/genetic a/TICdb)[15]. All of these fusion sequences were undertaken for BLAT study (genome.ucsc.edu) and only fusion sequences showing 100% sequence similarity scores, specifically with the two partner chromosomes, were taken for further analysis. Thousand base pair sequences i.e. 500 base pairs upstream and downstream (Breakpoint Island [BpIs]) flanking the breakpoint of each partner chromosome was retrieved from UCSC genome browser (genome. ucsc.edu) [16] (Table S1). Genomic sequence of GAPDH housekeeping gene preferably from core exonic and intron exon flanking region was taken from the same as control, where translocation is not evident.
Computational Analysis
The analysis of BpIs (Fig. 3) was initiated by exploring the SDs in flanking regions of breakpoints. Along with this, it was also analyzed whether breakpoints falls in intronic or exonic region for presence of repeats using UCSC Genome Browser. Further, the recombination signal sequences and Stress Induced Duplex Destabilization sites (SIDDs) were studied by RSS database [17] (http://www.itb.cnr.it/rss/), and WebSIDD server [18] (http://orange.genomecenter.ucdavis. edu /benham /sidd/) respectively in the BpIs. The resultant SIDD region sequences were further evaluated for their physico-chemical characteristics and GC content by means of diproGB [19] (http://diprogb.fli-leibniz.de/) and DNA base composition analysis tool (http://molbiol-tools.ca/Jie_ Zheng/) respectively.
RESULT
In silico Multiparametric investigation of BpIs was done in order to find out underlying mechanisms and assess the association of genomic features which could be correlated with the breakpoints. To achieve this, each partner chromosome was examined for presence of SDs, Repetitive elements (Alus), Genes, RSS, SIDD sites at the breakpoint and BpIs. The sequences exhibiting destabilized regions were further analyzed for Flexibility, Stability, Stacking Energy and AT Content.
Prevalence of SDs, Repeats and RSS in BpIss
We checked BpIs flanking region for presence of duplications, by gradually increasing the window at a regular interval of 2,000-bp in order to eliminate the risk of missing some of the duplicated segments (Fig. 4). The result represents that SDs are mapped within distance of 0.01 to 3 Mbs in the flanking regions of breakpoints either proximally or distally, for all translocations considered in this study. Their genomic locations were identified using the tracks "Segmental Dups" from UCSC genome browser (Table 1). The study of breakpoint junction sequences confirmed that all 5’ and 3’ breakpoints are located within intronic regions of the respective gene (Table S1). The Repeat Masker track in the UCSC genome browser was used to determine repetitive DNA elements in the breakpoints and BpIs. Analysis showed that, all translocations have been found to be flanked by Alu sequences in both or either of translocation partner chromosome. Other repeats like MIRs, LINE, LTR DNA elements, low complexity and simple repeats were also present in proximity of breakpoint region (Table S2). Breakpoint regions demonstrated higher occurrence of Alu repeats showing an increase varying from 2.1 to 6.5 folds as compared to their total Alu density in respective chromosomes (Fig. 5A1, A2). By utilizing RIC (Recombination Information Content) algorithm, we gained a local view of potent cRSSs within all translocation breakpoints (Fig. 5A1, A2). The cRSS with the highest ex vivo recombination potential reached a “pass” value for RSS12 with RIC ≥-38.81, while RSS23 with RIC ≥-58.45 have been found by RIC threshold. In total, we recognized more than twenty five cRSS of 12-bp and 23-bp spacer with the highest RIC score -27.35 and -52.11 respectively. Predominantly, cRSS at the BpIs was found to be higher in numbers for chromosome 21 as compared to others (Table S2).
Location of segmental duplicons in flanking regions of breakpoints.
| S No. | Translocations | Chr. Partner with Cytoband Location of SDs | Genomic Locations of SDs(feb 2009/hg19) | ˜ Distance from Breakpoint (Mbs) | Position from Breakpoint |
|---|---|---|---|---|---|
| 1) | t(2;2)(p21;p23.2) | 2p22.3 | 36218226-36219966 | 0.6 | U |
| 2p22.3 | 36216160-36218225 | 0.7 | D | ||
| 2) | t(10;10)(q21.2;q11.21) | 10q21.1 | 58185254-58208638 | 0.3 | U |
| 10q11.22 | 46687877-46704002 | 0.3 | D | ||
| 3) | t(12;15)(p13.2;q25.3) | 12p13.2 | 10374384-10375699 | 0.1 | D |
| 15q26.1 | 90890819-90892143 | 0.02 | U | ||
| 4) | t(5;6)(q32;q22.1) | 5q32 | 146085501-146087235 | 0.03 | D |
| 6q23.2 | 134617874-134619908 | 1.6 | U | ||
| 5a) | t(21;21)(q22.2;22.3) | 21q22.3 | 44009044-44010518 | 0.1 | U |
| 21q22.3 | 44007565-44009039 | 0.4 | U | ||
| 5b) | t(21;21)(q22.2;22.3) | 21q22.3 | 44009044-44010518 | 0.1 | U |
| 21q22.3 | 44007565-44009039 | 0.4 | U | ||
| 6) | t(7;15)(p21.2;q21.1) | 15q21.1 | 44896399-44898146 | 0.1 | D |
| 7p21.2 | 14978252-14979981 | 0.1 | U | ||
| 7) | t(X;1)(p11.23;p34.3) | 1p34.2 | 43355720-43357999 | 0.7 | U |
| Xp11.4 | 40694053-40697053 | 0.8 | D | ||
| 8) | t(5;8)(p13.1;q12.1) | 5p14.2 | 23299576-23305432 | 1.5 | D |
| 8q12.1 | 59332721-59339423 | 0.2 | U | ||
| 9) | t(12;3)(q14.3;p14.2) | 12q13.3 | 56904826-56906972 | 1.0 | D |
| 3p22.2 | 36808134-36810280 | 2.3 | D | ||
| 10a) | t(12;9)(q14.3;p23) | 12q22 | 93277561-93278745 | 2.6 | U |
| 9p24.1 | 4944404-4945893 | 0.9 | U | ||
| 10b) | t(12;9)(q14.3;p23) | 12q22 | 93277561-93278745 | 2.6 | U |
| 9p24.1 | 4944404-4945893 | 0.9 | U | ||
| 11) | t(9;15)(q34.2;q14) | 9q34.13 | 135894808-135896554 | 0.1 | D |
| 15q21.3 | 53229042-53230780 | 1.8 | U | ||
| 12) | t(21;22)(q22.2;q12.2) | 22q13.33 | 51226859-51244566 | 2.3 | U |
| 21q22.3 | 48100446-48117693 | 0.9 | U | ||
| 13a) | t(11;22)(q24.3;q12.2) | 22q13.2 | 43172344-43173365 | 1.4 | U |
| 11q23.3 | 118431342-118432360 | 1.0 | D | ||
| 13b) | t(11;22)(q24.3;q12.2) | 22q13.2 | 43172344-43173365 | 1.4 | U |
| 11q23.3 | 118431342-118432360 | 1.0 | D |
Incidence of Destabilization Sites, AT Percentage and Other Physico-Chemical Properties at BpIss
SIDD site can be described as compilation of successive base pairs whose free energy values (G(x)) are <4.0 kcal/mole, considered as the threshold for region as being destabilized which is evaluated by WebSIDD server with default parameters. Our result reflects that at least one destabilization site is there for each chromosome partner of all translocations whilst sequence from chromosome 9 shows maximum numbers of destabilization sites within BpIs (Table S2). Representative destabilization profile plots are shown below (Fig. 5B1, B2). On the whole, result of AT(%) cotent was found to be lower for the BpIs as compared with highest destabilized region successively when base composition analysis was performed for the particular chromosomal translocation (Fig. 5F1, F2) (Table S3). DiProGB analysis server was utilized in order to identify the BpIs & particular destabilized regions with respect to their dinucleotide properties and results are plotted (Fig. 5C-E). Result was constant in all translocations where stability showed decreasing trends while flexibility index and stacking energy showed increasing trend as compared to expected values (Table S3).
Analysis of Control Gene Sequence
Additionally GAPDH control gene sequences were examined for the above parameters other than SDs for comparison as control. Our results confirmed absolute absence of RSS, Repeats, and SIDD sites whilst Flexibility, stability, stacking energy values and AT% could distinguish the breakpoint and non-breakpoint region based on cutoff values derived from ROC curve analysis as depicted in Table S4.
DISCUSSION
Development of Innovative and sophisticated technologies for genome sequencing ended up in identifying Gene fusions as molecular signature in broad range of solid tumours, which were initially considered only to be associated with hematological tumours [20-25]. In order to explicate the cause of these chromosomal breaks, numerous potential biological mechanisms such as DNA repair by non-homologous end joining (NHEJ), Alu arbitrated homologous recombination, illegitimate V(D)J recombination and various others have been suggested [26, 27]. For acquisition of in- depth knowledge pertaining to these molecular mechanisms, there is requisite to punctuate that whether there is direct link between the particular pattern of local genomic sequence and breakpoint regions which may provide us with the clue for the cellular processes that promote chromosome rearrangements [12, 28, 29].
Substantial attention regarding the importance of Segmental Duplications in genetic dis-orders has been revealed due to significant advances in molecular cytogenetics field [30, 31]. Earlier studies describes the possible mechanisms of direct involvement of low copy repeats (LCRs) or SDs in the occurrence of t(11;22) [32, 33]. Interestingly, our bio-informatic analysis, for first time demonstrates the existence of SDs flanking the breakpoint regions of solid tumor translocations and their potential role in rearrangements of DNA segments. These duplicons are found to be placed across several mega-bases, either present at common breakpoint regions or elsewhere in same sub chromosomal region. These closest duplicons are homologous sequences that search for and anchor with each other, thereby making the recombination a feasible event showing their ability to serve as substrate for aberrant genomic rearrangement.
Though, the enormous role of particular genomic architectures has been already established as casual mechanism of recurrent rearrangements but till date, this information is only confined to individual translocation cases. Hence, to the best of our knowledge, this study is a novel approach in terms of analyzing all the parameters (that may play crucial role in chromosome break) collectively for many translocations of epithelial tumour.
Enhanced evidences for the incidence of high densities of repetitive DNA sequences like Alu repeats, at translocation breakpoint regions has proposed that these sequences act as hot spots for events of recombination and thus facilitates translocation process [33-35]. Our study also revealed presence of Alu repeats in the BpIs of all translocations amongst which chromosome 22 demonstrates the highest density (82.7%) whereas control gene under investigation exhibited complete absence of such repeats favoring the verity that Alu core sequence are of prime importance in promoting DNA strand exchange and genomic rearrangement.
It is distinctly apparent from this in silico analysis that, the breakpoint junctions are situated within the intronic portion of genomic sequences. This is indicative of the fact that the presence of breakpoints in the non-coding regions will not influence the functionality of the fusion gene so produced, highlighting the point that these intronic regions so present within the genome are purposeful. Our scrutiny affirmed the preponderance of SIDD sites in BpIs or at breakpoint junction but not in control genomic sequences. This articulates that regulatory genomic sites and recombination hot spots are more prone to stress driven strand separation which has a variety of inference in replication mechanism or transcriptional regulation [36, 37]. In addition, our result for all the translocation represents the existence of discrete characteristic - RSS, at or near the breakpoint region at least in either of the partner. Immense similarity is observed amongst the sequences of genes involved in solid tumour translocations and an authentic RSS sequences that normally comprises of at least a CAC, which is indispensable for RAG cleavage by V(D)J recombination thus making a double-strand break [38, 39]. On the other hand, control sequences including core exonic region and intron/exon boundary showed the nonappearance of RSS, SIDD which is again in support of actuality that there is a need of exceptional phenomena for chromosomal break leading to translocation other than mechanism of splicing.
Evaluation of comprehensive data concerned to physico-chemical features depicts inverse proportionality of stability with that of flexibility and stacking energy. This noticeably imply that base stacking and helical flexibility influences protein-DNA interactions to greater extent which therefore impact upon chromatin structure and generates genomic instability at the breakpoint region responsible for chromosome breakage [40-44]. Furthermore, comparatively high AT content is witnessed in the SIDD region as compared to whole BpIs which authenticate that AT Island are thermodynamically destabilized, exclusively flexible and remarkably prone to super helical stress thus enhancing the vulnerability of genomic breakage [45-47]. More to the point, perceptible discrimination amidst the breakpoint and non-breakpoint island with noteworthy specificity and sensitivity is observed, as per the cutoff values derived by ROC curve for Tm, Flexibility, AT content and stacking energy.
The in-depth knowledge derived from our computational assessment construed a straight correlation between high values stacking energy, flexibility, AT content with the presence of SIDD sites. Rest of the dynamic features though indispensable separately but are signifying discrepancy when compared with each other. Thus, these imputes are unswerving with the belief that presence of peculiar genomic patterns at or nearby breakpoints may act as driving force for DSBs causing translocations which has been illustrated in the CIRCOS plot (Fig. S2).
CONCLUSION
In summary, this multi-parametric bio-informatics methodology employed for analysis of genomic sequences of breakpoint coordinates furnishes us with superior perceptive of molecular mechanisms of DNA strand break during translocation leading to epithelial carcinogenesis. First and foremost, SDs in direct orientations are typically found close to breakpoints which might be obligatory for occurrence of efficacious recombination. Second, SIDD can be considered as a “Driver” mechanism that will help to distinguish a breakpoint region in any unidentified sequence owing to its direct connection with the physico chemical parameters as manifested from our research. Moreover, not any of the precise chromatin organization and DNA architectural motif revealed common signatures, though they are discretely imperative in increasing propensity for repeated cross over events. As our in silico investigations goes in analogue with the earlier reported in-vitro studies which eventually lead to authentication of the computational protocol followed here, reflecting the fact that chromosomal rearrangements are non-random actions which make DNA susceptible to DSBs in a precise and definite pattern. Inclusion of intron/exon flanking genomic sequences in control gene analysis strengthen the postulation that break involving translocation is inimitable by nature which is governed by manifold parameters like SIDD region, repeat, flexibility/stability, RSS, stacking energy and AT content. Furthermore this multi-parametric study can lead to the conceptualization of an algorithmic program which may predict possible breakpoint in any given sequence of human genome.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGEMENT
We would like to acknowledge The Gujarat Cancer & Research Institute for providing administrative support.
